- Antecedentes
- Etiología
- Epidemiología
- La vitamina K1 (filoquinona), una importante vitamina dietética, es producida por las plantas y las algas y está presente en grandes cantidades en las verduras de hoja verde. Junto con las vitaminas A, D y E, la vitamina K1 es una vitamina liposoluble.
- La flora microbiana intestinal normal produce vitamina K2 (menaquinonas).
- Se ha derivado una forma sintética de la vitamina K como agente farmacológico con mayor solubilidad en agua y que se designa como vitamina K3 (menadiona).
La deficiencia combinada de los factores de coagulación dependientes de la vitamina K (VKCFD) surge sólo en raras ocasiones como una condición congénita. La VKCFD congénita se hereda como una de las dos variantes; ambas son trastornos hemorrágicos autosómicos recesivos poco frecuentes. La presentación clínica más común de la actividad deficiente combinada de los factores de coagulación II (FII), FVII, FIX y FX (y las proteínas C, S y Z) se produce debido a la deficiencia adquirida de vitamina K, en particular como resultado de una enfermedad hepática, malabsorción o desnutrición, o asociada a una serie de medicamentos, sobre todo la sobredosis de warfarina o derivados de la misma.
La función de cada una de las proteínas dependientes de la vitamina K (VKDP) depende de la carboxilación gamma (Ƴ) normal de una serie de residuos de ácido glutámico en la región aminoterminal de las proteínas (el dominio Gla).1 Esta reacción requiere el ciclo normal de la vitamina K desde su forma reducida al estado oxidado, seguido de la regeneración de la vitamina K reducida.2, 3 Estas reacciones se llevan a cabo mediante la acción de dos enzimas del retículo endoplásmico (RE): la Ƴ-glutamil carboxilasa (GGCX) y la subunidad del complejo de la vitamina K 2,3 epóxido reductasa1 (VKORC1). Las dos variantes de la VKCFD surgen por defectos en los genes que codifican estas dos proteínas y no por defectos intrínsecos de los factores de coagulación. La actividad deficiente de GGCX da lugar a la VKCFD1, mientras que la actividad deficiente de VKORC1 da lugar a la VKCFD2. La presentación clínica más común de la VKCFD es la hemorragia grave durante la infancia, aunque el grado de deficiencia es muy variable y la presentación de la VKCFD puede producirse durante toda la infancia y en la edad adulta.4,5 Además, cada vez se reconoce más que la subcarboxilación y la función deficiente del VKDP que afecta a sistemas biológicos ajenos a la coagulación es común en la VKCFD, es muy variable en su presentación y puede presentar características clínicas considerablemente más tarde en la vida que el fenotipo hemorrágico, con implicaciones para la vigilancia y el manejo de la VKCFD.
McMillan y Roberts describieron el primer caso de hemorragia clínica resultante de una deficiencia congénita de Ƴ-carboxilación en 1966.6,7 La niña de un embarazo sin complicaciones manifestó hematomas a partir de la primera semana de vida, con exudación serosanguinolenta recurrente del muñón umbilical durante los primeros meses de vida. En el momento de la presentación, su tiempo de protrombina (TP) era de 95 segundos y su tiempo parcial de tromboplastina (TTP) era de 305 segundos. Al mezclar el plasma de la paciente en una proporción de 1:1 con plasma normal, cada una de estas pruebas se corrigió completamente hasta alcanzar el rango normal y, clínicamente, su hemorragia umbilical se detuvo tras una única infusión de plasma fresco congelado (PFC). Mientras que su recuento de plaquetas, fibrinógeno y FV y FVIII eran normales, la actividad de FII, FVII, FIX y FX estaban todos por debajo del límite inferior de detección (<3%). Tras la instauración de un programa de terapia diaria de vitamina K1 enteral, el niño creció y se desarrolló con normalidad, con sólo una leve tendencia a los hematomas. Una interrupción de la terapia con vitamina K1 durante un período de 9 días dio lugar a la reaparición de marcados hematomas graves.
Durante el medio siglo posterior, los diagnósticos de VKCFD han sido extremadamente raros. La colección más completa de casos notificados de VKCFD1 (defecto genético de GGCX asociado a factores de coagulación VKDP bajos) incluye menos de 30 familias; 5,7 se han descrito cuatro familias con VKCFD2.8 El grado de deficiencia es variable entre los casos notificados. La combinación particular de FII, FVII, FIX y FX deficientes da lugar a múltiples niveles de generación de trombina defectuosa. La actividad del sustrato protrombina es en sí misma deficiente, y además la generación de trombina a través del Factor Tisular/FVIIa y a través del complejo tenasa está alterada. La deficiencia de proteínas en cada una de las vías intrínseca, extrínseca y común de la cascada de la coagulación se refleja en la prolongación de las pruebas de detección del TP y del aPTT. La prolongación del TP puede ser más marcada que la del aPTT en los casos más leves. La carboxilación de Ƴ es necesaria también para la función normal de las proteínas anticoagulantes C, S y Z; sin embargo, la presentación común y las complicaciones graves de la VKCFD son resultado de hemorragias más que de trombosis.9
La etiología completa de la VKCFD quedó clara tras la clonación y caracterización del gen que codifica la proteína GGCX por Stafford y sus colegas en 199110,11 Stafford y sus colegas y Oldenburg y sus colegas en 2004.12,13,14 Estudios pedigríes posteriores han llevado a la designación de las dos variantes: VKCFD1, resultante de mutaciones en el gen de la Ƴ-glutamil carboxilasa (GGCX), y VKCFD2, resultante de mutaciones en el gen del complejo VKOR (VKORC1, o VKOR).15,16,17
La vitamina K existe en 3 formas:
Todas las proteínas dependientes de la vitamina K contienen un dominio Gla rico en ácido carboxiglutámico, y el papel de la vitamina K como cofactor en la correcta modificación postraduccional de este dominio es crítico para su función normal. Además de las proteínas implicadas en la coagulación, se aprecia cada vez más que una variedad de procesos biológicos se ven afectados por las proteínas dependientes de la vitamina K (VKDP) y la expresión fenotípica de la subcarboxilación puede estar asociada a manifestaciones hemorrágicas y no hemorrágicas. (Véase «Manifestaciones clínicas».) Además de la implicación de las proteínas anticoagulantes C y S en la inflamación, la acción de la proteína rica en Gla (GRP) en los monocitos/macrófagos está implicada en la interacción entre inflamación y calcificación. El VKDP osteocalcina, derivado de los osteoblastos, participa en el metabolismo de la glucosa y está asociado a la formación normal de los huesos y a su resistencia. Otros VKDP están implicados en la regulación de la proliferación celular (proteína del gen 6 específico de la detención del crecimiento, Gas6) y en la inhibición de la calcificación vascular y la mineralización de los tejidos (proteína Gla de la matriz, MGP), mientras que la función de varios VKDP adicionales se desconoce actualmente, incluidas las proteínas Gla transmembrana (TMG) y las proteínas Gla ricas en prolina (PRGP).2,18, 19, 20
Los factores de coagulación II, VII, IX y X y las proteínas C, S y Z contienen entre 9 y 13 glutamatos (Glu) en la secuencia de aminoácidos de sus respectivos dominios Gla. La modificación postraduccional insuficiente de estos glutamatos a ácido Ƴ-carboxiglutámico (Gla) es el paso enzimático que es defectuoso en ambas variantes de la VKCFD. La carboxilación del dominio Gla confiere una conformación dependiente del calcio que es crítica para la interacción normal de las proteínas con los fosfolípidos (por ejemplo, con la superficie fosfolipídica de la plaqueta activada) o con las células endoteliales. La GGCX es la proteína que realiza esta modificación postraduccional; sin embargo, la vitamina K en su forma reducida también es necesaria para actuar como cofactor. La vitamina K reducida se convierte en epóxido de vitamina K en el curso de la reacción, y la forma reducida debe ser regenerada para ciclos adicionales de catálisis a través de la acción de VKOR. El anticoagulante warfarina se opone al VKOR, de modo que un individuo que toma warfarina tiene una regeneración alterada de la vitamina K reducida, con un resultado controlado similar a la deficiencia de vitamina K o VKCFD: exceso de subcarboxilación y deficiencia funcional de FII, FVII, FIX y FX. 2,3 Los ciclos de la vitamina K pueden visualizarse como se muestra en la Figura 1.
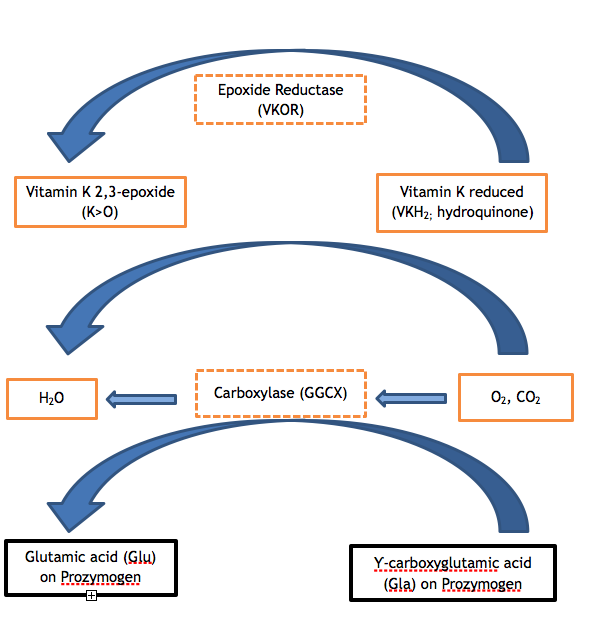
Figura 1. El ciclo de la vitamina K. La carboxilación de los residuos de ácido glutámico de las proteínas zimógenas dependientes de la vitamina K es necesaria para su correcto funcionamiento. La carboxilasa (GGCX) añade una molécula de dióxido de carbono a Glu para formar Gla en una reacción para la que la vitamina K es un cofactor esencial. La forma reducida de la vitamina K (hidroquinona) se oxida en el proceso, produciendo vitamina K 2,3-epóxido. La vitamina K reducida debe regenerarse a partir del epóxido mediante la acción enzimática del VKOR, de modo que la vitamina K reducida esté disponible para servir de cofactor en las reacciones de carboxilación posteriores. La actividad deficiente de cualquiera de las enzimas VKOR o GGCX (véase los recuadros sombreados de la figura) da lugar a la VKCFD.
De esta discusión se desprende que, aunque los VKDP se sintetizan y secretan normalmente, la función de la proteína es anormal como resultado de una carboxilación postraduccional insuficiente.
La purificación de GGCX y VKORC1, que son proteínas integrales de membrana, ha sido un reto y las estructuras cristalinas de estas proteínas aún no están resueltas. Sin embargo, está surgiendo una comprensión de los dominios funcionales de estas proteínas.1 Este esfuerzo se complementa con los recientes esfuerzos por correlacionar los defectos genéticos que afectan a regiones específicas de las enzimas con la gravedad y el espectro de la actividad deficiente de VKDP, tanto en el sistema de coagulación como en otros procesos fisiológicos. 5,8,21,22
El gen GGCX del brazo p del cromosoma 2 codifica una proteína transmembrana de 758 aminoácidos (AA) que se localiza en la membrana del retículo endoplásmico. La porción N-terminal de la enzima es citoplásmica. Le siguen cinco dominios transmembrana localizados en la membrana lipídica del RE. La porción C-terminal de la proteína es intraluminal en el RE. La región intraluminal contiene dominios hidrofóbicos importantes para interactuar con la vitamina K. Un importante dominio funcional (propuesto como AA 491-507) interactúa con el propéptido del VKDP, mientras que otra región de unión (AA 393-404) para el glutamato del VKDP facilita la gamma-carboxilación. De hecho, la primera correlación del fenotipo clínico de la VKCFD con una mutación en el gen GGCX describió una sustitución de leucina por arginina en la posición 394 de la región de unión al glutamato.15 Aunque la secuencia de los cuatro primeros dominios transmembrana de la GGCX está muy conservada evolutivamente, se desconoce su función en humanos. La proteína descrita aquí se expresa a partir del gen GGCX completo. Una isoforma 2 del ARNm de la GGCX que carece del exón 2 (Δ2GGCX) puede codificar una proteína que conserva la actividad enzimática, aunque la importancia fisiológica de esta isoforma no está determinada (véase también «Herencia y mutaciones causales»).23
El gen VKOR del cromosoma 16 codifica una proteína transmembrana de 163 AA. La topología básica de la membrana de la proteína VKOR es controvertida: por ejemplo, se sigue debatiendo si la proteína tiene tres o cuatro dominios transmembrana, y si el sitio activo altamente conservado de VKOR C132XXC135 para el ataque nucleofílico a la vitamina K es citoplasmático o está dentro del lumen del RE. Una importante región estructural que involucra las partes de arginina en AA 98 y 100 constituye un motivo de retención de ER de di-arginina que es necesario para mantener VKOR localizado en el sitio requerido de actividad. Una única mutación de VKOR (VKORC1p.Arg98Trp) representa todos los casos de VKCFD2, por lo que no existe la oportunidad de que las correlaciones genotipo/fenotipo informen sobre la comprensión de las interacciones enzimáticas funcionales de la reductasa (de forma análoga a las posibilidades que existen con VKCFD1). 1,8,24,25
Existe la posibilidad de que aún no se hayan identificado genes adicionales que modifiquen la acción de estas proteínas. Sin embargo, los datos de los animales sugieren que no existe una vía independiente y redundante de la carboxilasa en todos los tejidos, a la luz de la letalidad embrionaria tanto de los ratones sin GGCX como sin VKOR.26,27 Cuando se identificó la proteína VKOR, Rost y sus colegas identificaron una proteína paralógica denominada VKOR-like 1 (VKORL1), que es capaz de reducir el K>O;13 se ha sugerido un papel de VKORL1 para rescatar alguna función de la vitamina K en tejidos extrahepáticos durante la anticoagulación con antagonistas de la vitamina K.1 No hay datos que sugieran que VKORL1 tenga un papel fisiológico significativo en el apoyo a la carboxilación hepática de los factores de coagulación, por lo que no se cree que la proteína paralógica sea relevante para la VKCFD en este momento.
La VKCFD es muy rara. La colección más completa de casos publicados de VKCFD1 (en la que está presente una mutación genética en GGCX en asociación con la disminución de la actividad de los factores de coagulación) incluye 33 individuos afectados (26 probandos por separado).5 No se ha descrito ninguna influencia de la raza o la etnia, y se han notificado casos de África, Asia, Europa, América del Norte y América del Sur.7,21,28 La VKCFD2 es más rara, con 10 casos notificados de cuatro familias de origen caucásico norteamericano, alemán, italiano y libanés.8