- Hintergrund
- Etiologie
- Epidemiologie
Der kombinierte Mangel an Vitamin-K-abhängigen Gerinnungsfaktoren (VKCFD) tritt nur selten als angeborene Erkrankung auf. Kongenitale VKCFD wird als eine von zwei Varianten vererbt; beide sind seltene autosomal rezessive Blutungsstörungen. Die häufigere klinische Präsentation einer kombinierten mangelhaften Aktivität der Gerinnungsfaktoren II (FII), FVII, FIX und FX (und der Proteine C, S und Z) tritt aufgrund eines erworbenen Vitamin-K-Mangels auf, insbesondere infolge einer Lebererkrankung, Malabsorption oder Mangelernährung, oder in Verbindung mit einer Reihe von Medikamenten, vor allem einer Überdosierung von Warfarin oder Warfarinderivaten.
Die Funktion jedes der Vitamin-K-abhängigen Proteine (VKDP) hängt von der normalen Gamma (Ƴ)-Carboxylierung einer Reihe von Glutaminsäureresten im aminoterminalen Bereich der Proteine (der Gla-Domäne) ab.1 Diese Reaktion erfordert einen normalen Zyklus von Vitamin K von seiner reduzierten Form in den oxidierten Zustand, gefolgt von einer Regeneration des reduzierten Vitamin K.2, 3 Diese Reaktionen werden durch die Wirkung zweier Enzyme des endoplasmatischen Retikulums (ER) erreicht: die Ƴ-Glutamylcarboxylase (GGCX) und die Vitamin K 2,3-Epoxidreduktase-Komplex-Untereinheit1 (VKORC1). Die beiden Varianten der VKCFD entstehen durch Defekte in den Genen, die diese beiden Proteine kodieren, und nicht durch intrinsische Defekte in den Gerinnungsfaktoren. Mangelnde Aktivität von GGCX führt zu VKCFD1, während mangelnde Aktivität von VKORC1 zu VKCFD2 führt. Die häufigste klinische Präsentation von VKCFD sind schwere Blutungen im Säuglingsalter, obwohl der Grad des Mangels sehr variabel ist und die Präsentation von VKCFD während der gesamten Kindheit und im Erwachsenenalter auftreten kann.4,5 Darüber hinaus wächst die Erkenntnis, dass eine Untercarboxylierung und mangelhafte Funktion von VKDP, die biologische Systeme außerhalb der Gerinnung betrifft, bei VKCFD häufig vorkommt, in der Präsentation sehr variabel ist und klinische Merkmale deutlich später im Leben als der Blutungsphänotyp auftreten können, was Auswirkungen auf die Überwachung und das Management von VKCFD hat.
McMillan und Roberts beschrieben 1966 den ersten Fall einer klinischen Blutung als Folge einer angeborenen Ƴ-Carboxylierungsstörung.6,7 Das weibliche Kind einer unkomplizierten Schwangerschaft zeigte Blutergüsse, die im Alter von einer Woche begannen, mit rezidivierendem serosanguinösem Nässen aus dem Nabelstumpf in den ersten Lebensmonaten. Bei der Vorstellung lag ihre Prothrombinzeit (PT) bei 95 Sekunden und ihre partielle Thromboplastinzeit (PTT) bei 305 Sekunden. Nach dem Mischen des Plasmas der Patientin im Verhältnis 1:1 mit normalem Plasma korrigierte sich jeder dieser Tests vollständig in den Normalbereich und klinisch stoppte ihre Nabelblutung nach einer einzigen Infusion von frischem gefrorenem Plasma (FFP). Während ihre Thrombozytenzahl, Fibrinogen und FV und FVIII normal waren, lag die Aktivität von FII, FVII, FIX und FX alle unter der unteren Nachweisgrenze (<3%). Nach Einleitung einer täglichen enteralen Vitamin-K1-Therapie wuchs und entwickelte sich das Kind normal mit nur einer leichten Neigung zu Blutergüssen. Eine Unterbrechung der Vitamin-K1-Therapie über einen Zeitraum von 9 Tagen führte zur Rückkehr ausgeprägter schwerer Blutergüsse.
In dem darauffolgenden halben Jahrhundert waren Diagnosen von VKCFD extrem selten. Die umfangreichste Sammlung gemeldeter Fälle von VKCFD1 (genetischer Defekt von GGCX in Verbindung mit niedrigen VKDP-Gerinnungsfaktoren) umfasst weniger als 30 Sippen; 5,7 vier Familien mit VKCFD2 wurden beschrieben.8 Der Grad des Mangels ist bei den gemeldeten Fällen unterschiedlich. Die spezielle Kombination von defektem FII, FVII, FIX und FX führt zu einer mehrfach gestörten Thrombinbildung. Die Aktivität des Substrats Prothrombin ist selbst defizitär, und zusätzlich ist die Thrombinbildung sowohl über den Gewebefaktor/FVIIa als auch über den Tenase-Komplex beeinträchtigt. Der Mangel an Proteinen in jedem der intrinsischen, extrinsischen und gemeinsamen Wege der Gerinnungskaskade spiegelt sich in der Verlängerung der Screening-Tests PT und aPTT wider. Die Verlängerung der PT kann in leichteren Fällen stärker ausgeprägt sein als die Verlängerung der aPTT. Die Ƴ-Carboxylierung ist auch für die normale Funktion der gerinnungshemmenden Proteine C, S und Z erforderlich; die häufige Präsentation und die schweren Komplikationen der VKCFD resultieren jedoch eher aus Blutungen als aus Thrombosen.9
Die vollständige Ätiologie der VKCFD wurde nach der Klonierung und Charakterisierung des Gens, das für das GGCX-Protein kodiert, durch Stafford und Kollegen im Jahr 199110,11 und Oldenburg und Kollegen im Jahr 2004 klar.12,13,14 Nachfolgende Stammbaumstudien haben zur Benennung der zwei Varianten geführt: VKCFD1, resultierend aus Mutationen im Ƴ-Glutamylcarboxylase-Gen (GGCX), und VKCFD2, resultierend aus Mutationen im VKOR-Komplex-Gen (VKORC1, oder VKOR).15,16,17
Vitamin K existiert in 3 Formen:
- Vitamin K1 (Phyllochinon), ein wichtiges Nahrungsvitamin, wird von Pflanzen und Algen produziert und ist in großen Mengen in grünem und Blattgemüse enthalten. Zusammen mit den Vitaminen A, D und E ist Vitamin K1 ein fettlösliches Vitamin.
- Die normale mikrobielle Darmflora produziert Vitamin K2 (Menachinone).
- Eine synthetische Form von Vitamin K wurde als pharmakologischer Wirkstoff mit größerer Wasserlöslichkeit abgeleitet und als Vitamin K3 (Menadion) bezeichnet.
Die Vitamin-K-abhängigen Proteine enthalten alle eine Ƴ-Carboxyglutaminsäure-reiche Gla-Domäne, und die Rolle von Vitamin K als Co-Faktor bei der korrekten posttranslationalen Modifikation dieser Domäne ist entscheidend für ihre normale Funktion. Zusätzlich zu den Proteinen, die an der Gerinnung beteiligt sind, wird zunehmend erkannt, dass eine Vielzahl von biologischen Prozessen von Vitamin-K-abhängigen Proteinen (VKDP) beeinflusst werden und dass die phänotypische Ausprägung der Untercarboxylierung mit blutenden und nicht-blutenden Manifestationen verbunden sein kann. (Siehe „Klinische Manifestationen“.) Neben der Beteiligung der gerinnungshemmenden Proteine C und S an der Entzündung ist die Wirkung des Gla-reichen Proteins (GRP) in Monozyten/Makrophagen am Zusammenspiel von Entzündung und Verkalkung beteiligt. Das von Osteoblasten stammende VKDP Osteocalcin ist am Glukosestoffwechsel beteiligt und mit normaler Knochenbildung und Knochenfestigkeit assoziiert. Weitere VKDP sind an der Regulation der Zellproliferation (Growth Arrest Specific gene 6 protein, Gas6) und an der Hemmung der Gefäßverkalkung und der Gewebemineralisierung (Matrix-Gla-Protein, MGP) beteiligt, während die Funktion einiger weiterer VKDP derzeit noch unbekannt ist, darunter Transmembran-Gla-Proteine (TMG) und prolinreiche Gla-Proteine (PRGP).2,18, 19, 20
Die Gerinnungsfaktoren II, VII, IX und X sowie die Proteine C, S und Z enthalten zwischen 9 und 13 Glutamate (Glu) in der Aminosäuresequenz ihrer jeweiligen Gla-Domäne. Eine unzureichende posttranslationale Modifikation dieser Glutamate zu Ƴ-Carboxyglutaminsäure (Gla) ist der enzymatische Schritt, der in beiden Varianten der VKCFD defekt ist. Die Carboxylierung der Gla-Domäne verleiht eine kalziumabhängige Konformation, die für die normale Interaktion der Proteine mit Phospholipiden (z. B. mit der Phospholipidoberfläche des aktivierten Blutplättchens) oder mit Endothelzellen entscheidend ist. Das GGCX ist das Protein, das diese posttranslationale Modifikation durchführt; es wird jedoch auch Vitamin K in seiner reduzierten Form benötigt, um als Kofaktor zu wirken. Reduziertes Vitamin K wird im Verlauf der Reaktion in Vitamin-K-Epoxid umgewandelt, und die reduzierte Form muss für weitere Katalysezyklen durch die Wirkung von VKOR regeneriert werden. VKOR wird durch das Antikoagulans Warfarin bekämpft, so dass bei einer Person, die Warfarin einnimmt, die Regeneration von Vitamin K reduziert beeinträchtigt ist, mit einem kontrollierten Ergebnis, das einem Vitamin-K-Mangel oder einer VKCFD ähnelt: ein Überschuss an untercarboxyliertem und funktionell defizientem FII, FVII, FIX und FX. 2,3 Die Vitamin-K-Zyklen können wie in Abbildung 1 dargestellt visualisiert werden.
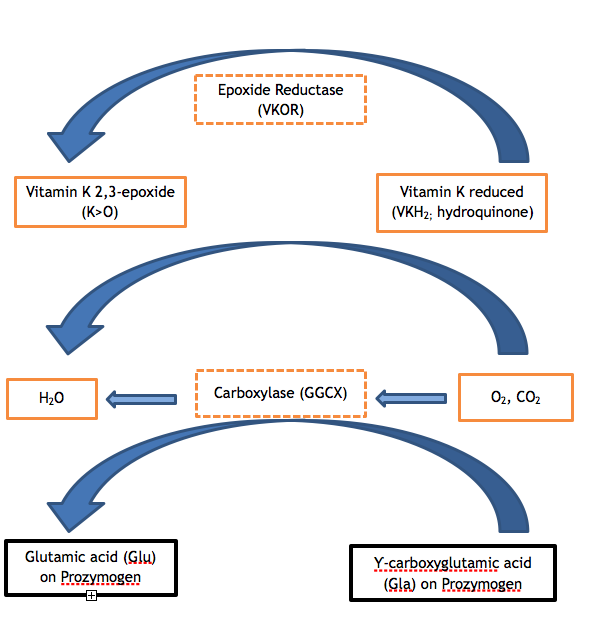
Abbildung 1. Der Vitamin-K-Zyklus. Die Carboxylierung von Glutaminsäureresten der zymogenen Vitamin-K-abhängigen Proteine ist für deren Funktion notwendig. Die Carboxylase (GGCX) fügt ein Kohlendioxidmolekül an Glu an, um Gla in einer Reaktion zu bilden, für die Vitamin K ein essentieller Co-Faktor ist. Die reduzierte Form des Vitamin K (Hydrochinon) wird dabei oxidiert, es entsteht Vitamin K 2,3-Epoxid. Vitamin K reduziert muss durch die enzymatische Wirkung von VKOR aus dem Epoxid regeneriert werden, damit Vitamin K reduziert als Cofaktor für nachfolgende Carboxylierungsreaktionen zur Verfügung steht. Mangelnde Aktivität eines der Enzyme VKOR oder GGCX (siehe schraffierte Kästchen in der Abbildung) führt zu VKCFD.
Aus dieser Diskussion geht hervor, dass VKDPs zwar normal synthetisiert und sezerniert werden, die Funktion des Proteins jedoch aufgrund einer unzureichenden posttranslationalen Carboxylierung gestört ist.
Die Reinigung von GGCX und VKORC1, die integrale Membranproteine sind, war eine Herausforderung und die Kristallstrukturen dieser Proteine sind noch nicht aufgelöst. Nichtsdestotrotz zeichnet sich ein Verständnis der funktionellen Domänen dieser Proteine ab.1 Diese Bemühungen werden ergänzt durch die jüngsten Bemühungen, Gendefekte, die bestimmte Regionen der Enzyme betreffen, mit der Schwere und dem Spektrum der mangelhaften VKDP-Aktivität zu korrelieren, sowohl im Gerinnungssystem als auch in anderen physiologischen Prozessen. 5,8,21,22
Das GGCX-Gen auf dem p-Arm von Chromosom 2 kodiert für ein 758 Aminosäuren (AA) großes Transmembranprotein, das an der Membran des endoplasmatischen Retikulums lokalisiert ist. Der N-terminale Teil des Enzyms ist zytoplasmatisch. Es folgen fünf Transmembrandomänen, die in der Lipidmembran des ER lokalisiert sind. Der C-terminale Teil des Proteins befindet sich intralumenal im ER. Die intralumenale Region enthält hydrophobe Domänen, die für die Interaktion mit Vitamin K wichtig sind. Eine wichtige funktionelle Domäne (vorgeschlagen als AA 491-507) interagiert mit dem Propeptid des VKDP, während eine andere Bindungsregion (AA 393-404) für das VKDP-Glutamat die Gamma-Carboxylierung erleichtert. Tatsächlich beschrieb die erste Korrelation des klinischen Phänotyps von VKCFD mit einer Mutation im GGCX-Gen eine Substitution von Leucin durch Arginin an Position 394 der Glutamat-Bindungsregion.15 Obwohl die Sequenz der ersten vier GGCX-Transmembrandomänen evolutionär hoch konserviert ist, ist ihre Funktion beim Menschen nicht bekannt. Das hier beschriebene Protein wird von dem GGCX-Gen in voller Länge exprimiert. Eine Isoform 2 der GGCX-mRNA, der das Exon 2 fehlt (Δ2GGCX), kodiert möglicherweise ein Protein, das die enzymatische Aktivität beibehält, obwohl die physiologische Bedeutung dieser Isoform nicht bestimmt ist (siehe auch „Vererbung und ursächliche Mutationen“).23
Das VKOR-Gen auf Chromosom 16 kodiert ein 163 AA großes Transmembranprotein. Die grundlegende Membrantopologie des VKOR-Proteins ist umstritten: So wird z. B. diskutiert, ob das Protein drei oder vier Transmembrandomänen besitzt und ob die hochkonservierte VKOR-C132XXC135-Aktivstelle für den nukleophilen Angriff auf Vitamin K zytoplasmatisch oder im ER-Lumen liegt. Eine wichtige strukturelle Region umfasst die Arginin-Anteile an AA 98 und 100, die ein Di-Arginin-ER-Retentionsmotiv bilden, das notwendig ist, um VKOR an der erforderlichen Aktivitätsstelle lokalisiert zu halten. Eine einzige VKOR-Mutation (VKORC1p.Arg98Trp) macht alle Fälle von VKCFD2 aus, so dass es keine Möglichkeit für Genotyp/Phänotyp-Korrelationen gibt, um das Verständnis der funktionellen enzymatischen Interaktionen der Reduktase zu informieren (analog zu den Möglichkeiten, die bei VKCFD1 bestehen). 1,8,24,25
Es besteht die Möglichkeit, dass zusätzliche Gene, die die Wirkung dieser Proteine modifizieren, noch nicht identifiziert sind. Tierexperimentelle Daten deuten jedoch darauf hin, dass ein unabhängiger, redundanter Carboxylase-Weg nicht in allen Geweben existiert, wenn man die embryonale Letalität sowohl von GGCX-Knockout als auch von VKOR-Knockout bei Mäusen betrachtet.26,27 Zum Zeitpunkt der Identifizierung des VKOR-Proteins identifizierten Rost und Kollegen ein paraloges Protein mit der Bezeichnung VKOR-like 1 (VKORL1), das in der Lage ist, K>O zu reduzieren;13 eine Rolle für VKORL1 zur Rettung einiger Vitamin-K-Funktionen in extrahepatischen Geweben während der Antikoagulation mit Vitamin-K-Antagonisten wurde vorgeschlagen.1 Es gibt keine Daten, die darauf hindeuten, dass VKORL1 eine signifikante physiologische Rolle bei der Unterstützung der hepatischen Carboxylierung von Gerinnungsfaktoren spielt, so dass das parologe Protein derzeit nicht als relevant für die VKCFD angesehen wird.
VKCFD ist sehr selten. Die umfangreichste Sammlung veröffentlichter Fälle von VKCFD1 (bei der eine Genmutation in GGCX in Verbindung mit einer verminderten Aktivität der Gerinnungsfaktoren vorliegt) umfasst 33 betroffene Personen (26 verschiedene Probanden).5 Es wurde kein Einfluss von Rasse oder ethnischer Zugehörigkeit beschrieben, und es wurden Fälle aus Afrika, Asien, Europa, Nordamerika und Südamerika berichtet.7,21,28 VKCFD2 ist seltener, mit 10 berichteten Fällen aus vier Familien nordamerikanisch-kaukasischer, deutscher, italienischer und libanesischer Herkunft.8