- Sfondo
- Eziologia
- Epidemiologia
Carenza combinata di fattori di coagulazione vitamina K-dipendenti (VKCFD) si presenta solo raramente come una condizione congenita. Il VKCFD congenito è ereditato come una delle due varianti; entrambi sono rari disturbi autosomici recessivi dell’emorragia. La presentazione clinica più comune di un’attività carente combinata dei fattori di coagulazione II (FII), FVII, FIX e FX (e delle proteine C, S e Z) si verifica a causa di una carenza acquisita di vitamina K, in particolare derivante da malattie epatiche, malassorbimento o malnutrizione, o associata a una serie di farmaci, in particolare il sovradosaggio di warfarin o derivati del warfarin.
La funzione di ciascuna delle proteine vitamina K-dipendenti (VKDP) dipende dalla normale carbossilazione gamma (Ƴ) di un certo numero di residui di acido glutammico nella regione amino-terminale delle proteine (il dominio Gla).1 Questa reazione richiede il normale passaggio della vitamina K dalla sua forma ridotta allo stato ossidato, seguito dalla rigenerazione della vitamina K ridotta.2, 3 Queste reazioni avvengono tramite l’azione di due enzimi del reticolo endoplasmatico (ER): la Ƴ-glutamil carbossilasi (GGCX) e il complesso della vitamina K 2,3 epossido reduttasi subunità1 (VKORC1). Le due varianti di VKCFD sorgono a causa di difetti nei geni che codificano queste due proteine e non a causa di difetti intrinseci nei fattori di coagulazione. L’attività carente di GGCX porta alla VKCFD1, mentre l’attività carente di VKORC1 porta alla VKCFD2. La presentazione clinica più comune di VKCFD è grave emorragia durante l’infanzia, anche se il grado di carenza è molto variabile e la presentazione di VKCFD può verificarsi durante l’infanzia e in età adulta.4,5 Inoltre, c’è un crescente riconoscimento del fatto che la sottocarbossilazione e la funzione carente di VKDP che colpisce i sistemi biologici al di fuori della coagulazione è comune nella VKCFD, è altamente variabile nella presentazione e può presentare caratteristiche cliniche molto più tardi nella vita rispetto al fenotipo di sanguinamento, con implicazioni per la sorveglianza e la gestione della VKCFD.
McMillan e Roberts hanno descritto il primo caso di emorragia clinica derivante da un deficit congenito di Ƴ-carbossilazione nel 1966.6,7 Il neonato femmina di una gravidanza senza complicazioni ha manifestato ecchimosi a partire da una settimana di età, con ricorrenti trasudazioni sierosanguinose dal moncone ombelicale durante i primi mesi di vita. Alla presentazione il suo tempo di protrombina (PT) era di 95 secondi e il suo tempo di tromboplastina parziale (PTT) era di 305 secondi. Mescolando il plasma della paziente in un rapporto 1:1 con plasma normale, ognuno di questi test è stato corretto completamente nel range normale e clinicamente il suo sanguinamento ombelicale si è fermato dopo una singola infusione di plasma fresco congelato (FFP). Mentre la sua conta delle piastrine, il fibrinogeno, il FV e il FVIII erano normali, l’attività di FII, FVII, FIX e FX erano tutti al di sotto del limite inferiore di rilevazione (<3%). All’istituzione di un programma di terapia enterale quotidiana con vitamina K1, il bambino è cresciuto e si è sviluppato normalmente con solo una lieve tendenza alla formazione di lividi. Un’interruzione della terapia con vitamina K1 per un periodo di 9 giorni ha portato al ritorno di gravi lividi marcati.
Durante il successivo mezzo secolo, le diagnosi di VKCFD sono state estremamente rare. La raccolta più completa dei casi riportati di VKCFD1 (difetto genetico di GGCX associato a bassi fattori di coagulazione VKDP) comprende meno di 30 parentele; 5,7 quattro famiglie con VKCFD2 sono state descritte.8 Il grado di carenza è variabile tra i casi riportati. La particolare combinazione di carenza di FII, FVII, FIX e FX si traduce in livelli multipli di generazione difettosa di trombina. L’attività del substrato protrombina è di per sé carente, e inoltre la generazione di trombina attraverso il fattore tissutale/FVIIa e attraverso il complesso tenasi è compromessa. La carenza di proteine in ciascuna delle vie intrinseche, estrinseche e comuni della cascata di coagulazione si riflette nel prolungamento dei test di screening PT e aPTT. Il prolungamento del PT può essere più marcato del prolungamento dell’aPTT nei casi più lievi. La carbossilazione Ƴ è necessaria anche per la normale funzione delle proteine anticoagulanti C, S e Z; tuttavia, la presentazione comune e le gravi complicazioni della VKCFD derivano da emorragie piuttosto che da trombosi.9
L’eziologia completa della VKCFD è diventata chiara dopo la clonazione e la caratterizzazione del gene che codifica la proteina GGCX da parte di Stafford e colleghi nel 199110,11 Stafford e colleghi e Oldenburg e colleghi nel 2004.12,13,14 Successivi studi di pedigree hanno portato alla denominazione delle due varianti: VKCFD1, risultante da mutazioni nel gene della Ƴ-glutamil carbossilasi (GGCX), e VKCFD2, risultante da mutazioni nel gene del complesso VKOR (VKORC1, o VKOR).15,16,17
La vitamina K esiste in 3 forme:
- La vitamina K1 (fillochinone), un’importante vitamina alimentare, è prodotta da piante e alghe ed è presente in grandi quantità nelle verdure verdi e a foglia. Insieme alle vitamine A, D ed E, la vitamina K1 è una vitamina liposolubile.
- La normale flora microbica intestinale produce la vitamina K2 (menachinoni).
- Una forma sintetica di vitamina K è stata derivata come agente farmacologico con una maggiore solubilità in acqua e designata vitamina K3 (menadione).
Le proteine vitamina K dipendenti contengono tutte un dominio Gla ricco di acido carbossiglutammico, e il ruolo della vitamina K come cofattore nella corretta modifica post-traslazionale di questo dominio è fondamentale per la loro normale funzione. Oltre alle proteine coinvolte nella coagulazione, è sempre più apprezzato che una varietà di processi biologici sono influenzati dalle proteine vitamina K dipendenti (VKDP) e l’espressione fenotipica della sotto-carbossilazione può essere associata a manifestazioni emorragiche e non. Oltre al coinvolgimento delle proteine anti-coagulanti C e S nell’infiammazione, l’azione della proteina ricca di Gla (GRP) nei monociti/macrofagi è coinvolta nell’interazione tra infiammazione e calcificazione. L’osteocalcina VKDP derivata dagli osteoblasti è coinvolta nel metabolismo del glucosio e associata alla normale formazione delle ossa e alla forza delle ossa. Altri VKDP sono coinvolti nella regolazione della proliferazione cellulare (Growth Arrest Specific gene 6 protein, Gas6) e nell’inibizione della calcificazione vascolare e della mineralizzazione dei tessuti (matrix Gla protein, MGP), mentre la funzione di molti altri VKDP è attualmente sconosciuta, comprese le proteine Gla transmembrana (TMG) e le proteine Gla ricche di prolina (PRGP).2,18, 19, 20
I fattori di coagulazione II, VII, IX e X e le proteine C, S e Z contengono da 9 a 13 glutammati (Glu) nella sequenza aminoacidica dei rispettivi domini Gla. L’insufficiente modifica post-traslazionale di questi glutammati in acido Ƴ-carbossiglutammico (Gla) è il passo enzimatico che è difettoso in entrambe le varianti di VKCFD. La carbossilazione del dominio Gla conferisce una conformazione calcio-dipendente che è critica per la normale interazione delle proteine con i fosfolipidi (ad esempio, con la superficie fosfolipidica della piastrina attivata) o con le cellule endoteliali. La GGCX è la proteina che esegue questa modifica post-traslazionale; tuttavia, anche la vitamina K nella sua forma ridotta è necessaria per agire come cofattore. La vitamina K ridotta viene convertita in epossido di vitamina K nel corso della reazione, e la forma ridotta deve essere rigenerata per ulteriori cicli di catalisi attraverso l’azione di VKOR. VKOR è contrastato dall’anticoagulante warfarin, così che un individuo che assume warfarin ha una rigenerazione alterata della vitamina K ridotta, con un risultato controllato simile alla carenza di vitamina K o VKCFD: eccesso di sottocarbossilato e funzionalmente carente di FII, FVII, FIX e FX. 2,3 I cicli della vitamina K possono essere visualizzati come mostrato nella Figura 1.
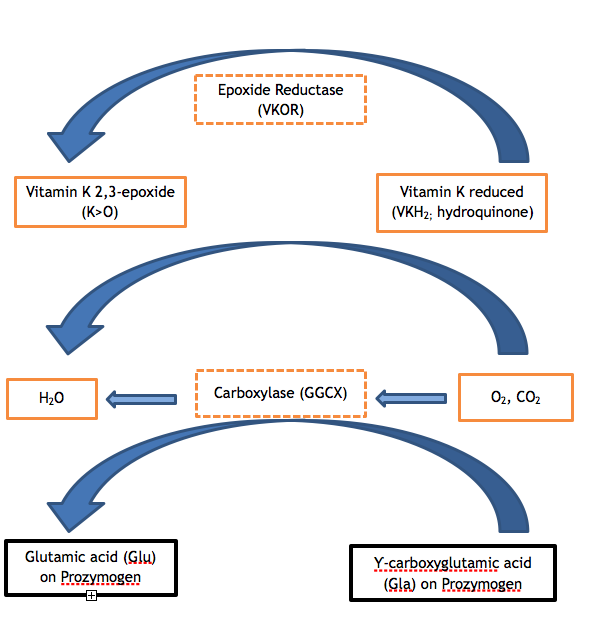
Figura 1. Il ciclo della vitamina K. La carbossilazione dei residui di acido glutammico delle proteine zimogene vitamina K-dipendenti è necessaria per il loro corretto funzionamento. La carbossilasi (GGCX) aggiunge una molecola di anidride carbonica a Glu per formare Gla in una reazione per la quale la vitamina K è un cofattore essenziale. La forma ridotta della vitamina K (idrochinone) è ossidata nel processo, producendo la vitamina K 2,3-epossido. La vitamina K ridotta deve essere rigenerata dall’epossido dall’azione enzimatica di VKOR in modo che la vitamina K ridotta sia disponibile per servire come cofattore per le successive reazioni di carbossilazione. Un’attività carente di uno dei due enzimi VKOR o GGCX (vedi le caselle tratteggiate nella figura) provoca la VKCFD.
È evidente da questa discussione che mentre i VKDP sono sintetizzati e secreti normalmente, la funzione della proteina è anormale come risultato di una carbossilazione post-traslazionale insufficiente.
La purificazione di GGCX e VKORC1, che sono proteine integrali di membrana, è stata difficile e le strutture cristalline di queste proteine non sono ancora risolte. Tuttavia, una comprensione dei domini funzionali di queste proteine sta emergendo.1 Questo sforzo è completato da recenti sforzi per correlare i difetti genici che interessano regioni specifiche degli enzimi con la gravità e lo spettro dell’attività VKDP carente, sia nel sistema di coagulazione che in altri processi fisiologici. 5,8,21,22
Il gene GGCX sul braccio p del cromosoma 2 codifica per una proteina transmembrana di 758 aminoacidi (AA) che è localizzata sulla membrana del reticolo endoplasmatico. La porzione N-terminale dell’enzima è citoplasmatica. Seguono cinque domini transmembrana, localizzati nella membrana lipidica dell’ER. La porzione C-terminale della proteina è intralumenale nell’ER. La regione intralumenale contiene domini idrofobici importanti per interagire con la vitamina K. Un importante dominio funzionale (proposto come AA 491-507) interagisce con il propeptide della VKDP, mentre un’altra regione di legame (AA 393-404) per il glutammato della VKDP facilita la gamma-carbossilazione. Infatti, la prima correlazione del fenotipo clinico della VKCFD con una mutazione nel gene GGCX ha descritto una sostituzione della leucina con l’arginina in posizione 394 della regione di legame del glutammato.15 Sebbene la sequenza dei primi quattro domini transmembrana GGCX sia altamente conservata a livello evolutivo, la loro funzione nell’uomo non è nota. La proteina qui descritta è espressa dal gene GGCX completo. Un’isoforma 2 dell’mRNA di GGCX che manca dell’esone 2 (Δ2GGCX) può codificare una proteina che mantiene l’attività enzimatica, sebbene il significato fisiologico di questa isoforma non sia determinato (vedi anche “Ereditarietà e mutazioni causali”).23
Il gene VKOR del cromosoma 16 codifica una proteina transmembrana di 163 AA. La topologia di membrana di base della proteina VKOR è controversa: per esempio, si continua a discutere se la proteina abbia tre o quattro domini transmembrana, e se il sito attivo altamente conservato VKOR C132XXC135 per l’attacco nucleofilo alla vitamina K sia citoplasmatico o all’interno del lume ER. Un’importante regione strutturale coinvolge le società di arginina a AA 98 e 100 che costituiscono un motivo di ritenzione ER di-arginina che è necessario per mantenere VKOR localizzato nel sito di attività richiesto. Una singola mutazione di VKOR (VKORC1p.Arg98Trp) rappresenta tutti i casi di VKCFD2, e quindi non esiste l’opportunità di correlazioni genotipo/fenotipo per informare la comprensione delle interazioni funzionali enzimatiche della reduttasi (analogamente alle possibilità che esistono con VKCFD1). 1,8,24,25
Esiste la possibilità che ulteriori geni che modificano l’azione di queste proteine possano essere ancora non identificati. I dati sugli animali suggeriscono che un percorso indipendente e ridondante di carbossilasi non esiste in tutti i tessuti, tuttavia, alla luce della letalità embrionale sia di GGCX knockout che di VKOR knockout nei topi.26,27 Al momento dell’identificazione della proteina VKOR, Rost e colleghi hanno identificato una proteina paraloga etichettata VKOR-like 1 (VKORL1), che è in grado di ridurre la K>O;13 è stato suggerito un ruolo per VKORL1 per salvare alcune funzioni della vitamina K in tessuti extra-epatici durante l’anticoagulazione con antagonisti della vitamina K.1 Non ci sono dati che suggeriscano che VKORL1 abbia un ruolo fisiologico significativo nel sostenere la carbossilazione epatica dei fattori della coagulazione, quindi la proteina paraloga non è ritenuta rilevante per la VKCFD in questo momento.
VKCFD è molto rara. La raccolta più completa di casi pubblicati di VKCFD1 (in cui è presente una mutazione genica in GGCX in associazione con una ridotta attività dei fattori della coagulazione) comprende 33 individui affetti (26 probandi separati).5 Non è stata descritta alcuna influenza della razza o dell’etnia, e sono stati riportati casi dall’Africa, dall’Asia, dall’Europa, dal Nord America e dal Sud America.7,21,28 La VKCFD2 è più rara, con 10 casi riportati da quattro famiglie di origine caucasica, tedesca, italiana e libanese del Nord America.8