- Tło
- Etiologia
- Epidemiologia
Zespół niedoboru czynników krzepnięcia zależnych od witaminy K (VKCFD) występuje rzadko jako stan wrodzony. Wrodzony VKCFD jest dziedziczony jako jeden z dwóch wariantów; oba są rzadkimi, dziedziczonymi autosomalnie recesywnie zaburzeniami krwawienia. Częściej spotykana postać kliniczna łącznego niedoboru aktywności czynników krzepnięcia II (FII), FVII, FIX i FX (oraz białek C, S i Z) występuje z powodu nabytego niedoboru witaminy K, w szczególności wynikającego z chorób wątroby, zaburzeń wchłaniania lub niedożywienia, lub związanego z przyjmowaniem wielu leków, zwłaszcza przedawkowaniem warfaryny lub jej pochodnych.
Funkcja każdego z białek zależnych od witaminy K (VKDP) zależy od prawidłowej gamma (Ƴ)-karboksylacji pewnej liczby reszt kwasu glutaminowego w aminokońcowym regionie białek (domena Gla).1 Reakcja ta wymaga normalnego cyklu witaminy K z jej zredukowanej postaci do stanu utlenionego, po którym następuje regeneracja zredukowanej witaminy K.2, 3 Reakcje te zachodzą dzięki działaniu dwóch enzymów retikulum endoplazmatycznego (ER): karboksylazy Ƴ-glutamylowej (GGCX) i podjednostki kompleksu reduktazy witaminy K 2,3 epoksydowej1 (VKORC1). Dwa warianty VKCFD powstają w wyniku defektów w genach kodujących te dwa białka, a nie w wyniku wewnętrznych defektów czynników krzepnięcia. Deficyt aktywności GGCX powoduje powstanie VKCFD1, natomiast deficyt aktywności VKORC1 prowadzi do powstania VKCFD2. Najczęstszym objawem klinicznym VKCFD jest ciężki krwotok w okresie niemowlęcym, chociaż stopień niedoboru jest bardzo zróżnicowany i objawy VKCFD mogą wystąpić w całym dzieciństwie i w wieku dorosłym.4,5 Ponadto coraz częściej uznaje się, że niedokarboksylacja i upośledzona funkcja VKDP wpływająca na układy biologiczne poza krzepnięciem jest powszechna w VKCFD, jest wysoce zmienna w prezentacji i może prezentować cechy kliniczne znacznie później w życiu niż fenotyp krwawienia, co ma wpływ na nadzór i postępowanie w VKCFD.
McMillan i Roberts opisali pierwszy przypadek klinicznego krwotoku wynikającego z wrodzonego niedoboru karboksylacji Ƴ w 1966 r.6,7 U dziecka płci żeńskiej z niepowikłanej ciąży wystąpiło zasinienie rozpoczynające się w 1. tygodniu życia, z nawracającym wyciekiem krwi z kikuta pępowiny przez pierwsze miesiące życia. W chwili zgłoszenia czas protrombinowy (PT) wynosił 95 sekund, a czas częściowej tromboplastyny (PTT) 305 sekund. Po zmieszaniu osocza pacjentki w stosunku 1:1 z osoczem normalnym, każdy z tych testów skorygował się całkowicie do zakresu normy, a klinicznie krwawienie pępkowe ustało po pojedynczym wlewie świeżo mrożonego osocza (FFP). Podczas gdy liczba płytek krwi, fibrynogen, FV i FVIII były prawidłowe, aktywność FII, FVII, FIX i FX była poniżej dolnej granicy wykrywalności (<3%). Po rozpoczęciu programu codziennej terapii dojelitowej witaminą K1 dziecko rosło i rozwijało się normalnie, z jedynie łagodną tendencją do siniaków. Przerwanie terapii witaminą K1 na okres 9 dni spowodowało powrót znacznego nasilenia siniaków.
W ciągu następnych pół wieku rozpoznania VKCFD były niezwykle rzadkie. Najbardziej obszerny zbiór opisanych przypadków VKCFD1 (defekt genetyczny GGCX związany z niskim poziomem czynników krzepnięcia VKDP) obejmuje mniej niż 30 rodzin; 5,7 opisano cztery rodziny z VKCFD2.8 Stopień niedoboru jest różny w opisanych przypadkach. Szczególna kombinacja niedoboru FII, FVII, FIX i FX powoduje wielopoziomowe zaburzenia w wytwarzaniu trombiny. Aktywność protrombiny jako substratu jest upośledzona, a dodatkowo upośledzone jest wytwarzanie trombiny zarówno przez czynnik tkankowy/FVIIa, jak i przez kompleks tenazy. Niedobór białek w każdym z wewnątrzpochodnych, zewnątrzpochodnych i wspólnych szlaków kaskady krzepnięcia znajduje odzwierciedlenie w wydłużeniu zarówno testu przesiewowego PT, jak i aPTT. W łagodniejszych przypadkach wydłużenie PT może być bardziej wyraźne niż wydłużenie aPTT. Karboksylacja Ƴ jest również wymagana do prawidłowego funkcjonowania białek przeciwzakrzepowych C, S i Z, jednak częsta prezentacja i ciężkie powikłania VKCFD wynikają raczej z krwawienia niż z zakrzepicy.9
Pełna etiologia VKCFD stała się jasna po sklonowaniu i scharakteryzowaniu genu kodującego białko GGCX przez Stafforda i współpracowników w 1991 r.10,11 Stafford i współpracownicy oraz Oldenburg i współpracownicy w 2004 r.12,13,14 Kolejne badania rodowodowe doprowadziły do oznaczenia dwóch wariantów: VKCFD1, wynikającego z mutacji w genie karboksylazy Ƴ-glutamylowej (GGCX), oraz VKCFD2, wynikającego z mutacji w genie kompleksu VKOR (VKORC1, lub VKOR).15,16,17
Witamina K występuje w 3 formach:
- Witamina K1 (filochinon), ważna witamina dietetyczna, jest wytwarzana przez rośliny i algi i występuje w dużych ilościach w zielonych i liściastych warzywach. Wraz z witaminami A, D i E, witamina K1 jest witaminą rozpuszczalną w tłuszczach.
- Normalna flora bakteryjna jelit wytwarza witaminę K2 (menachinony).
- Syntetyczna forma witaminy K została uzyskana jako środek farmakologiczny o większej rozpuszczalności w wodzie i oznaczona jako witamina K3 (menadion).
Białka zależne od witaminy K zawierają domenę Gla bogatą w kwas karboksyglutaminowy Ƴ, a rola witaminy K jako kofaktora w prawidłowej modyfikacji potranslacyjnej tej domeny jest krytyczna dla ich prawidłowej funkcji. Oprócz białek biorących udział w krzepnięciu, coraz bardziej docenia się, że na wiele procesów biologicznych wpływają białka zależne od witaminy K (VKDP), a fenotypowa ekspresja niedokarboksylacji może być związana z objawami krwawienia i niekrwawienia. (Patrz „Objawy kliniczne.”) Oprócz udziału białek przeciwzakrzepowych C i S w zapaleniu, działanie białka bogatego w Gla (GRP) w monocytach/makrofagach jest zaangażowane w interakcję pomiędzy zapaleniem a zwapnieniem. Pochodząca z osteoblastów VKDP osteokalcyna jest zaangażowana w metabolizm glukozy i jest związana z prawidłowym tworzeniem kości i ich wytrzymałością. Inne VKDP biorą udział w regulacji proliferacji komórek (białko genu 6 specyficznego dla zahamowania wzrostu, Gas6) oraz w hamowaniu zwapnienia naczyń krwionośnych i mineralizacji tkanek (białko Gla macierzy, MGP), podczas gdy funkcja kilku innych VKDP jest obecnie nieznana, w tym transmembranowych białek Gla (TMG) i bogatych w prolinę białek Gla (PRGP).2,18, 19, 20
Czerwone czynniki krzepnięcia II, VII, IX i X oraz białka C, S i Z zawierają od 9 do 13 glutaminianów (Glu) w sekwencji aminokwasowej swoich domen Gla. Niewystarczająca potranslacyjna modyfikacja tych glutaminianów do kwasu Ƴ-karboksyglutaminowego (Gla) jest etapem enzymatycznym, który jest wadliwy w obu wariantach VKCFD. Karboksylacja domeny Gla nadaje zależną od wapnia konformację, która jest krytyczna dla normalnej interakcji tych białek z fosfolipidami (np. z powierzchnią fosfolipidową aktywowanej płytki) lub z komórkami śródbłonka. GGCX jest białkiem, które przeprowadza tę potranslacyjną modyfikację, jednak witamina K w zredukowanej postaci jest również wymagana do działania jako kofaktor. Zredukowana witamina K jest w trakcie reakcji przekształcana do epoksydu witaminy K, a forma zredukowana musi być regenerowana dla kolejnych cykli katalizy poprzez działanie VKOR. VKOR jest przeciwstawiany przez antykoagulant warfarynę, tak że osoba przyjmująca warfarynę ma upośledzoną regenerację zredukowanej witaminy K, z kontrolowanym wynikiem podobnym do niedoboru witaminy K lub VKCFD: nadmiar niedokarboksylowanych i funkcjonalnie wadliwych FII, FVII, FIX i FX. 2,3 Cykle witaminy K można zwizualizować w sposób przedstawiony na rycinie 1.
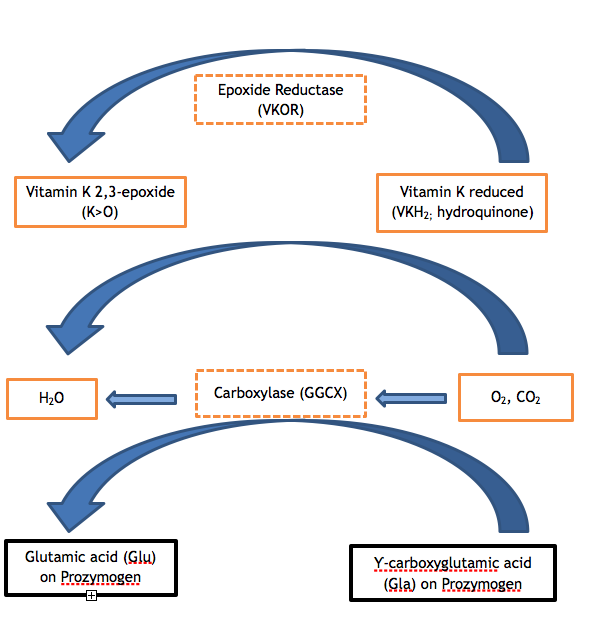
Figura 1. Cykl witaminy K. Karboksylacja reszt kwasu glutaminowego białek zymogennych zależnych od witaminy K jest niezbędna do ich prawidłowego funkcjonowania. Karboksylaza (GGCX) dodaje cząsteczkę dwutlenku węgla do Glu, tworząc Gla w reakcji, dla której witamina K jest niezbędnym kofaktorem. Zredukowana forma witaminy K (hydrochinon) jest w tym procesie utleniana, dając 2,3-epoksyd witaminy K. Zredukowana witamina K musi zostać zregenerowana z epoksydu przez enzymatyczne działanie VKOR, aby zredukowana witamina K mogła służyć jako kofaktor w kolejnych reakcjach karboksylacji. Niedostateczna aktywność któregokolwiek z enzymów VKOR lub GGCX (patrz zakreskowane pola na rysunku) powoduje VKCFD.
Z tej dyskusji wynika, że podczas gdy VKDP są syntetyzowane i wydzielane normalnie, funkcja białka jest nieprawidłowa w wyniku niewystarczającej karboksylacji potranslacyjnej.
Oczyszczanie GGCX i VKORC1, które są integralnymi białkami błonowymi, stanowiło wyzwanie, a struktury krystaliczne tych białek nie zostały jeszcze rozwiązane. Niemniej jednak, pojawia się zrozumienie funkcjonalnych domen tych białek.1 Wysiłki te są uzupełniane przez ostatnie wysiłki mające na celu skorelowanie defektów genów wpływających na specyficzne regiony enzymów z ciężkością i spektrum niedoboru aktywności VKDP, zarówno w układzie krzepnięcia, jak i w innych procesach fizjologicznych. 5,8,21,22
Gen GGCX na ramieniu p chromosomu 2 koduje 758 aminokwasowe (AA) białko transmembranowe, które jest zlokalizowane na błonie retikulum endoplazmatycznego. N-końcowa część enzymu jest cytoplazmatyczna. Następnie występuje pięć domen transmembranowych, zlokalizowanych w błonie lipidowej ER. C-końcowa część białka jest intralumenalna w ER. Region intralumenalny zawiera hydrofobowe domeny ważne dla interakcji z witaminą K. Ważna domena funkcjonalna (proponowana jako AA 491-507) oddziałuje z propeptydem VKDP, podczas gdy inny region wiążący (AA 393-404) dla glutaminianu VKDP ułatwia gamma-karboksylację. W rzeczywistości, pierwsza korelacja fenotypu klinicznego VKCFD z mutacją w genie GGCX opisywała zastąpienie leucyny przez argininę w pozycji 394 regionu wiążącego glutaminian.15 Chociaż sekwencja pierwszych czterech domen transmembranowych GGCX jest wysoce konserwowana ewolucyjnie, ich funkcja u ludzi nie jest znana. Opisywane tu białko ulega ekspresji z pełnej długości genu GGCX. Izoforma 2 mRNA GGCX pozbawiona eksonu 2 (Δ2GGCX) może kodować białko, które zachowuje aktywność enzymatyczną, chociaż fizjologiczne znaczenie tej izoformy nie zostało określone (patrz także „Dziedziczenie i mutacje przyczynowe”).23
Gen VKOR chromosomu 16 koduje białko transmembranowe o masie 163 AA. Podstawowa topologia błonowa białka VKOR jest kontrowersyjna: na przykład trwa dyskusja, czy białko ma trzy czy cztery domeny transmembranowe i czy wysoce konserwowane miejsce aktywne VKOR C132XXC135 dla ataku nukleofilowego na witaminę K jest cytoplazmatyczne czy znajduje się w świetle ER. Ważny region strukturalny obejmuj±cy cząsteczki argininy przy AA 98 i 100 tworzy di-arginininowy motyw retencyjny ER, który jest niezbędny do utrzymania VKOR w wymaganym miejscu aktywno¶ci. Pojedyncza mutacja VKOR (VKORC1p.Arg98Trp) odpowiada za wszystkie przypadki VKCFD2, a zatem nie ma możliwości uzyskania korelacji genotyp/fenotyp, które mogłyby przyczynić się do zrozumienia funkcjonalnych interakcji enzymatycznych reduktazy (analogicznie do możliwości istniejących w przypadku VKCFD1). 1,8,24,25
Istnieje możliwość, że dodatkowe geny, które modyfikują działanie tych białek mogą być jeszcze niezidentyfikowane. Dane z badań na zwierzętach sugerują, że niezależny, redundantny szlak karboksylazy nie istnieje we wszystkich tkankach, jednak w świetle śmiertelności embrionalnej zarówno nokautu GGCX, jak i nokautu VKOR u myszy.26,27 W czasie, gdy zidentyfikowano białko VKOR, Rost i współpracownicy zidentyfikowali paralogiczne białko oznaczone jako VKOR-like 1 (VKORL1), które jest zdolne do redukcji K>O;13 zasugerowano rolę VKORL1 w ratowaniu niektórych funkcji witaminy K w tkankach pozawątrobowych podczas antykoagulacji antagonistami witaminy K.1 Nie ma danych sugerujących, że VKORL1 odgrywa znaczącą rolę fizjologiczną we wspieraniu wątrobowej karboksylacji czynników krzepnięcia, dlatego uważa się, że to paralogiczne białko nie ma obecnie znaczenia dla VKCFD.
VKCFD występuje bardzo rzadko. Najbardziej obszerny zbiór opublikowanych przypadków VKCFD1 (w którym mutacja genu GGCX jest obecna w połączeniu ze zmniejszoną aktywnością czynników krzepnięcia) obejmuje 33 osoby dotknięte chorobą (26 odrębnych probantów).5 Nie opisano wpływu rasy ani pochodzenia etnicznego, a przypadki opisano w Afryce, Azji, Europie, Ameryce Północnej i Ameryce Południowej.7,21,28 VKCFD2 jest rzadsza, z 10 zgłoszonymi przypadkami z czterech rodzin pochodzenia północnoamerykańskiego, kaukaskiego, niemieckiego, włoskiego i libańskiego.8
W przypadku VKCFD2 nie opisano wpływu rasy ani pochodzenia etnicznego.